Find the location of transcriptional regulatory elements and transcription factoring binding using genomic data.
The gateway status and updates are here!
The dREG model in the gateway predicts the location of enhancers and promoters using PRO-seq, GRO-seq, or ChRO-seq data. The server takes as input bigWig files provided by the user, which represent PRO-seq signal on the plus and minus strand. The gateway uses a pre-trained dREG model to identify divergent transcript start sites and impute the predicted DNase-I hypersensitivity signal across the genome. The current dREG model works in any mammalian organism.
Registered users need only upload experimental data in the required format and push the start button. Once the job is finished, the user will be notified by e-mail. Results can be downloaded to the user’s local machine, or viewed in the Genome Browser via the handy trackhub link.
![]() Use the Danko lab's mapping pipeline (here) to prepare bigWig files from fastq files or convert BAM files of mapped reads to bigWig (here).
Use the Danko lab's mapping pipeline (here) to prepare bigWig files from fastq files or convert BAM files of mapped reads to bigWig (here).
![]() See our documentation, FAQ,
GitHub,
dREG paper, or
dREG protocol for additional questions.
See our documentation, FAQ,
GitHub,
dREG paper, or
dREG protocol for additional questions.
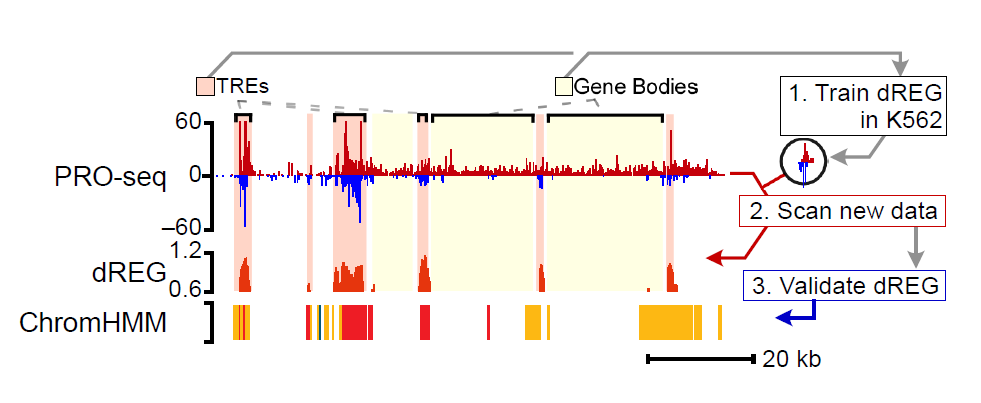
Click the figure to enlarge it
The dTOX models in the gateway predict the binding status of transcription factor binding sites using PRO-seq, ATAC-seq, or DNase-I-seq data. The server takes as input bigWig files provided by the user, which represent the PRO-seq, ATAC-seq, or DNase-1-seq signal on the plus and minus strand. The gateway uses two pre-trained dTOX models to identify transcription factor binding patterns genome-wide. The current dTOX models work in any mammalian organism and on any motif that has an associated position-weight matrix. To run the dTOX models on genomes other than hg19 and mm10, download the R package (here).
The web operations are same as the dREG model. Users need to login -> upload data -> run data. Results can be downloaded or viewed in the WashU Genome browser.
![]() Use the Danko lab's pipeline to convert BAM files of mapped reads to bigWig (here for PRO-seq), (here for DNase-I-seq), and (here for ATAC-seq).
Use the Danko lab's pipeline to convert BAM files of mapped reads to bigWig (here for PRO-seq), (here for DNase-I-seq), and (here for ATAC-seq).
![]() See our documentation, FAQ for additional questions. Currently the paper is in preparation.
See our documentation, FAQ for additional questions. Currently the paper is in preparation.
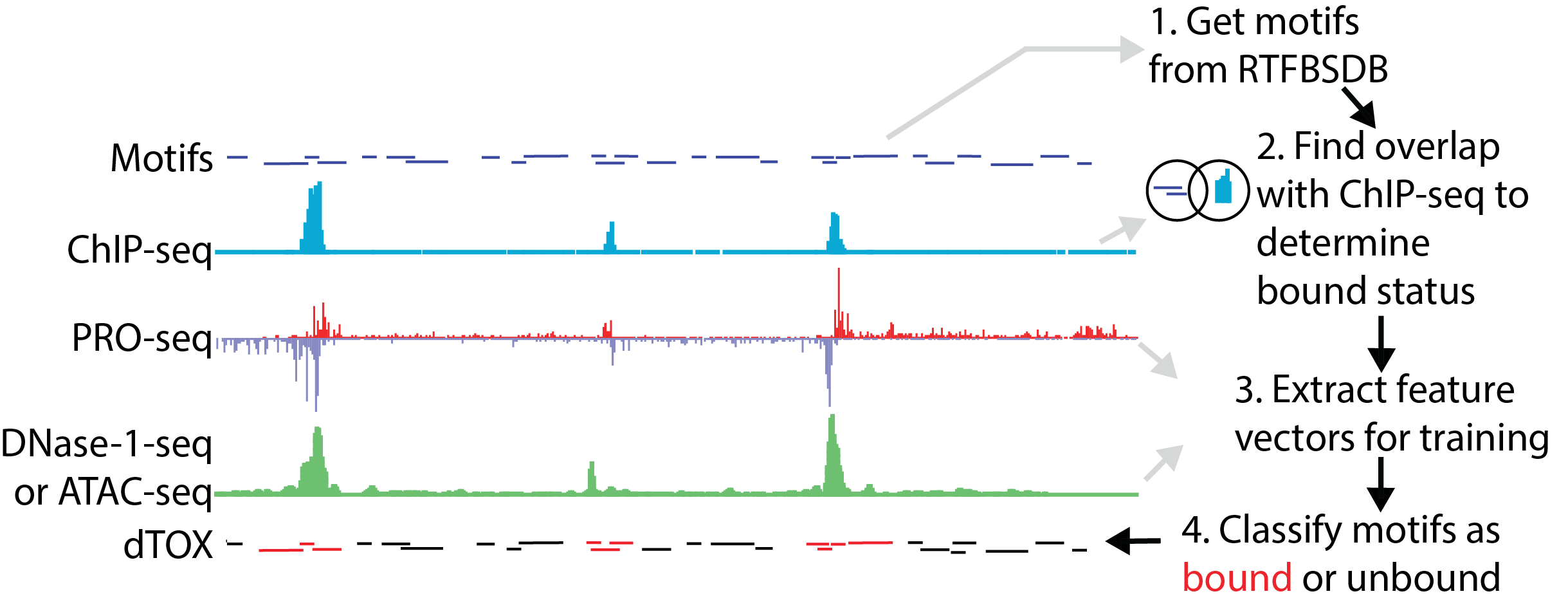
Click the figure to enlarge it
Transcription factors (TFs) regulate complex programs of gene transcription by binding to short DNA sequence motifs within transcription regulatory elements (TRE). tfTarge is a unified framework that identifies the "TF -> TRE -> target gene" networks that are differential regulated between two conditions, e.g. experimental vs. control, using PRO-seq/GRO-seq/ChRO-seq data as the input. The online service provies a convenient method for users without assuming knowledge with R environment, users can directly run the bigWig data on the dREG gateway.
Registered users need only upload experimental data in the required format and push the start button. Once the job is finished, the user will be notified by e-mail. Results can be downloaded to the user's local machine.
![]() Use the Danko lab's mapping pipeline (here) to prepare bigWig files from fastq files or convert BAM files of mapped reads to bigWig (here).
Use the Danko lab's mapping pipeline (here) to prepare bigWig files from fastq files or convert BAM files of mapped reads to bigWig (here).
![]() See our documentation, FAQ,
GitHub,
NG's paper, or
dREG protocol for additional questions.
See our documentation, FAQ,
GitHub,
NG's paper, or
dREG protocol for additional questions.
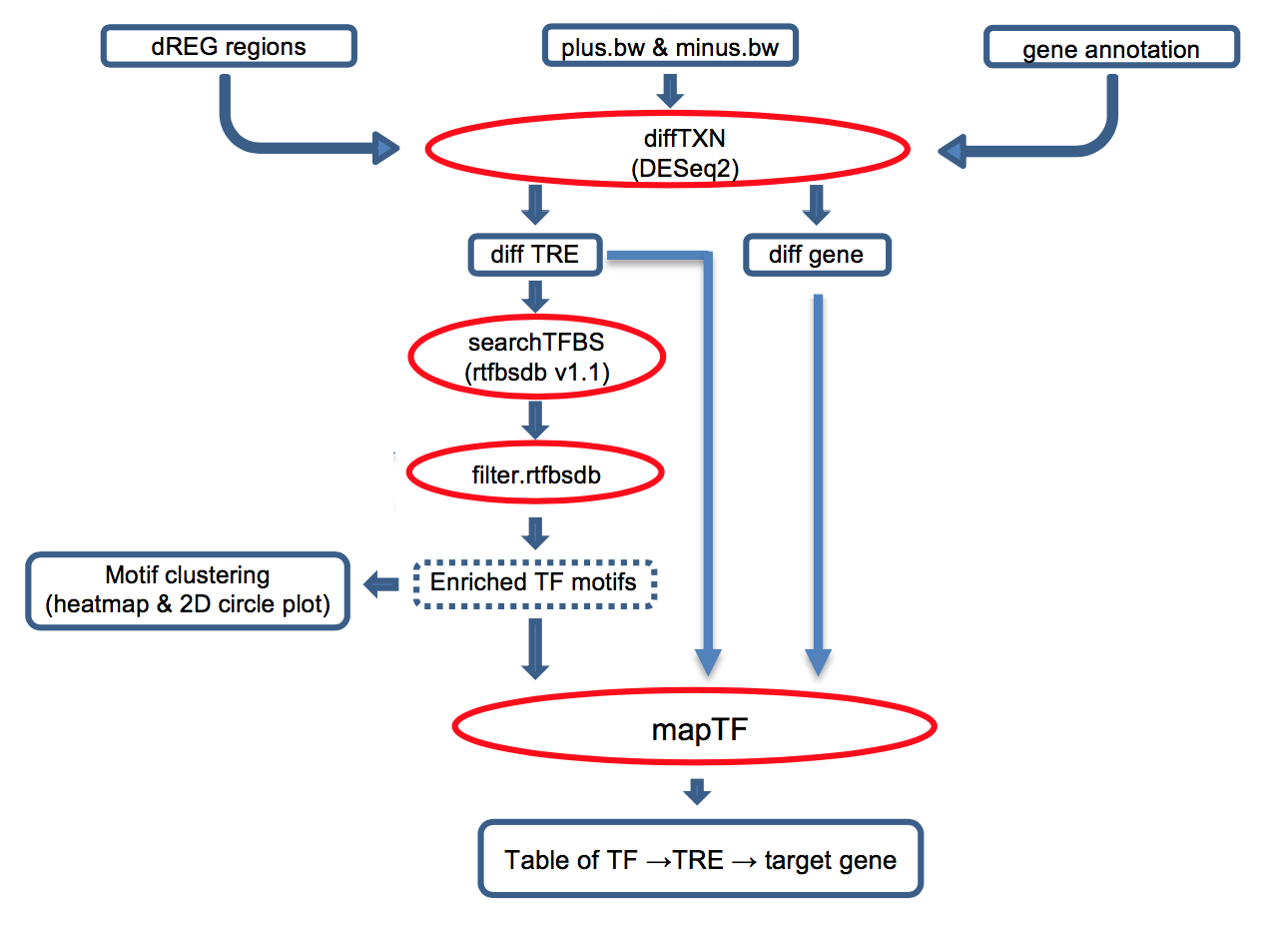
Click the figure to enlarge it
BayesPrism is a fully Bayesian inference of tumor microenvironment composition and gene expression. It consists of the deconvolution module and the embedding learning module. The deconvolution module leverages cell-type-specific expression profiles from scRNA-seq and implements a fully Bayesian inference to jointly estimate the posterior distribution of cell-type composition and cell type-specific gene expression from bulk RNA-seq expression of tumor samples. The embedding learning module uses Expectation-maximization (EM) to approximate the tumor expression using a linear combination of tumor pathways while conditional on the inferred expression and fraction of non-tumor cells estimated by the deconvolution module. Only the deconvolution module has been implemented as an online service due to the limitation of running time on the dREG gateway.
The web operations are the same as the dREG model. Users need to login -> upload data -> run data. Results can be downloaded and further analyzed in R or Python.
![]() See our documentation,
FAQ,
GitHub, or
paper
for additional questions.
See our documentation,
FAQ,
GitHub, or
paper
for additional questions.
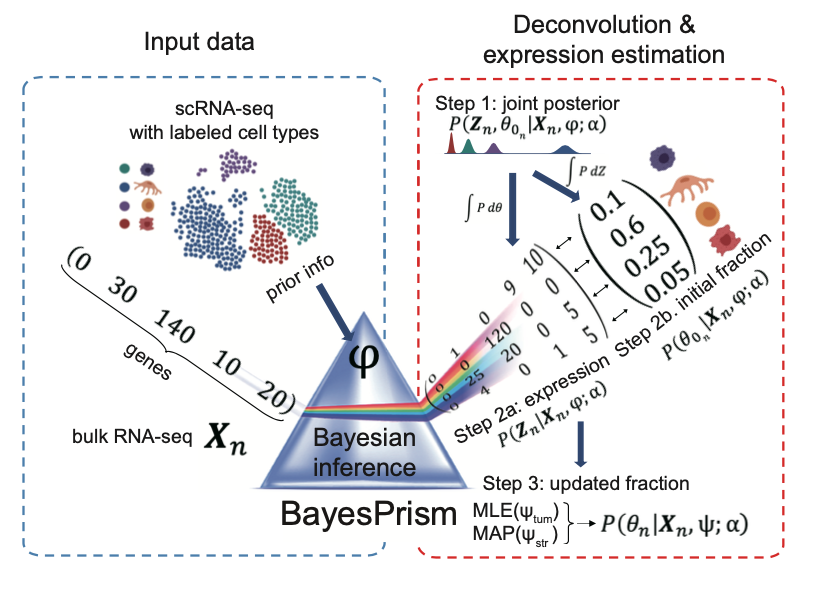
Click the figure to enlarge it
dHIT2 is a deep learning method for predicting histone modification marks. It can simultaneously transform primary transcripts (RO-seq) and DNA sequences into ChIP-seq profiles representing ten different histone modifications. The online service provies a convenient method for users without assuming knowledge with CUDA and python environment, users can directly run the bigWig data on the dHIT2 gateway.
The web operations are the same as the dREG model. Users need to login -> upload data -> run data. Results can be downloaded and further analyzed in Python.
![]() See our documentation,
FAQ
for additional questions.
See our documentation,
FAQ
for additional questions.
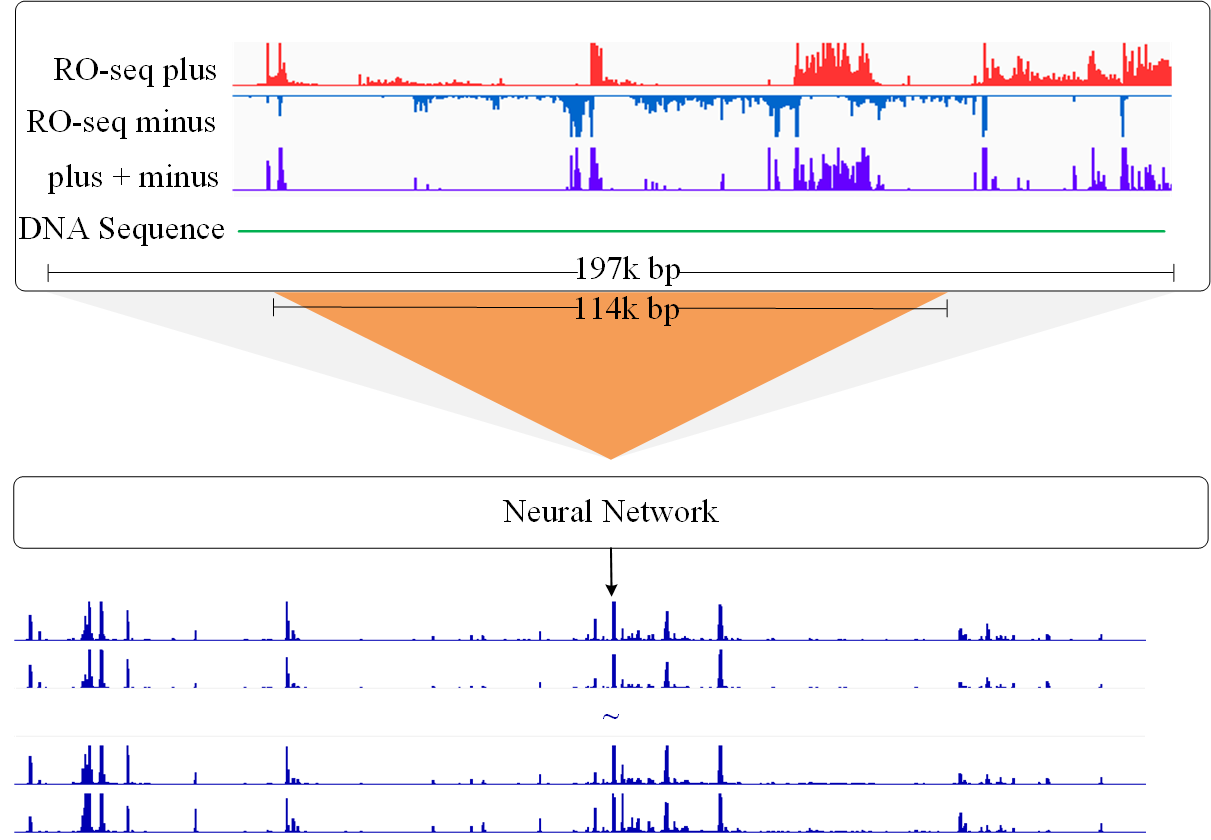
Click the figure to enlarge it
dHICA is a deep learning model that integrates DNA sequence data and chromatin accessibility to predict multiple levels of histone modifications (H3K122ac, H3K4me1, H3K4me2, H3K4me3, H3K27ac, H3K27me3, H3K36me3, H3K9ac, H3K9me3, and H4K20me1) simultaneously. There are two options for chromatin accessibility data: ATAC-seq (fold change over control) or DNase-seq (read-depth normalized signal).
The online service offers a user-friendly method for running dHICA without requiring knowledge of CUDA and python environments. Users can directly upload their chromatin accessibility data to the dHICA gateway for analysis.
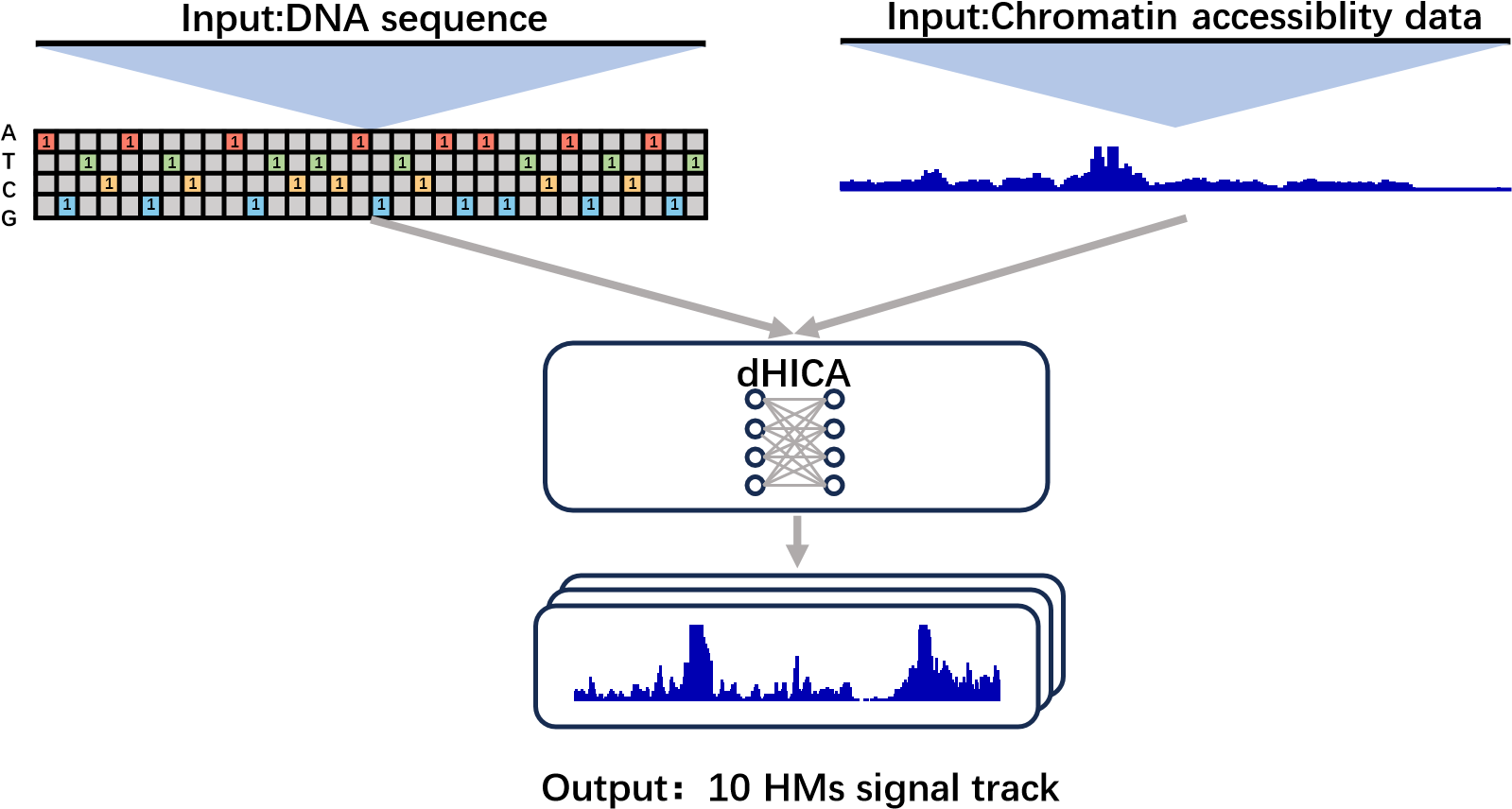
Click the figure to enlarge it
The dREG gateway is a cloud platform developed by the Danko lab at the Baker Institute, Cornell University and supported by the SciGap (Science Gateway Platform as a Service) and XSEDE (Extreme Science and Engineering Discovery Environment).
Currently, this gateway hosts four bioinformatics services for functional analysis of sequencing data, dREG peak calling, dTOX, tfTarget, and BayesPrism on XSEDE computing nodes. The architecture and details are here. |
Wang, N., Wang, Z., Danko, C. G., & Chu, T. (2022). Mapping Transcription Regulation with Run-on and Sequencing Data Using the Web-Based tfTarget Gateway. In DNA-Protein Interactions: Methods and Protocols (pp. 215-226). New York, NY: Springer US. |
 |
Chu, T., Wang, Z., Peer, D., & Danko, C. G. (2022). Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nature Cancer, 3, 505-517. |
 |
Wang, Z., Chu, T., Choate, L. A., & Danko, C. G. (2019). Identification of regulatory elements from nascent transcription using dREG. Genome Research, 29(2), 293-303. |
 |
Chu, T., Edward, J. R., Gregory, T. B., ... & Danko, C. G.(2018). Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme. Nature Genetics, 50, 1553-1564. |
 |
Danko, C. G., Hyland, S. L., Core, L. J., Martins, A. L., Waters, C. T., Lee, H. W., ... & Siepel, A. (2015). Identification of active transcriptional regulatory elements from GRO-seq data. Nature Methods, 12(5), 433-438. |
 |
Wang, Z., Chivu, A. G., Choate, L.A., ... & Danko, C. G. (2015). Prediction of histone post-translational modification patterns based on nascent transcription data. Nature Genetics, 54, 295-305. |
